
技術文章
Technical articles 更新時間:2025-07-23
更新時間:2025-07-23 點擊次數:179
點擊次數:179
《Advanced science》是Wiley出版集團旗下的一本開放獲取(Open Access)綜合性學術期刊,于2014年創刊。在中科院最新升級版分區表中,大類學科為材料科學1區,小類學科中化學綜合為1區、納米科技為2區、材料科學綜合為1區,屬于TOP期刊。《Advanced science》是一本跨學科開放獲取期刊,涵蓋材料科學、物理和化學、醫學和生命科學以及工程學等領域,主要發表這些領域的應用研究成果。
出版周期:12 issues/year;
影響因子:14.1;
ISSN:2198-3844;
發文量:3290篇/年;
審稿速度:平均12周;
版面費:USD 5270.00;

一、研究背景與目標
肺纖維化(PF)是多種肺部疾病的進展性致命結局,特發性肺纖維化(IPF)為主要亞型,由慢性肺部炎癥驅動,患者確診后平均生存期僅3-5年。全球發病率上升,新冠疫情后PF發生率達2-6%,約44.9%的新冠幸存者發展為PF。臨床僅吡非尼酮和尼達尼布獲批,療效有限,亟需闡明發病機制以開發新療法。成纖維細胞膠原合成是PF的核心特征,但其異常激活的觸發因素不明。本研究聚焦組織蛋白酶K(CTSK)在PF中的作用及機制。
二、關鍵技術總結
本文為探究組織蛋白酶 K(CTSK)在肺纖維化(PF)中的作用及機制,采用了多種實驗技術,涵蓋分子生物學、細胞生物學、動物模型及臨床樣本分析等領域,具體如下:
1. 肺纖維化模型:通過氣管內注射博來霉素(BLM)構建小鼠PF模型,用于評估CTSK對纖維化進展的影響;通過百草ku(PQ)誘導急性肺損傷(ALI)模型,結合不同時間點采樣進行蛋白質組學分析。
2. 基因編輯模型:構建成纖維細胞特異性CTSK條件敲除小鼠(Col1a2-CreERT;CTSK flox/flox),驗證內源性CTSK的作用。
3. 藥物干預:對模型小鼠施用重組CTSK(rCTSK)、CTSK抑制劑(odanacatib)、內吞抑制劑(Pitstop2)、SMAD3抑制劑(SIS3)及谷氨酰胺酶抑制劑(DON等),探究相關通路的作用。
4. 蛋白質組學與單細胞測序(scRNA-seq):通過蛋白質組學篩選PF模型小鼠肺組織中差異表達的組織蛋白酶家族成員,結合scRNA-seq(數據集 GSE111664、GSE136831 等)定位CTSK在成纖維細胞中的富集。
5. 細胞模型:使用人成纖維細胞系(MRC-5),經TGF-β1誘導模擬成纖維細胞-肌成纖維細胞轉化(FMT),研究CTSK對膠原合成的影響;分離原代小鼠肺成纖維細胞進行驗證。
6. 蛋白相互作用驗證:采用免疫共沉淀(Co-IP)驗證CTSK與SNX9的直接相互作用;通過AlphaFold3預測結合位點,結合丙氨酸突變實驗確定關鍵氨基酸殘基。
7. 分子表達檢測:利用Western blot檢測蛋白質表達水平,實時定量PCR(qRT-PCR)分析 mRNA 水平,免疫熒光染色觀察蛋白定位與共定位。
8. 非靶向與靶向代謝組學:通過LC-MS/MS對TGF-β1誘導的MRC-5細胞(經rCTSK處理)進行非靶向代謝組學分析,結合KEGG和MSEA富集分析篩選差異代謝通路(如谷氨酰胺代謝);通過靶向代謝組學檢測谷氨酰胺、谷氨酸及脯氨酸等代謝物水平。
9. 組織與病理分析:采用Masson三色染色和Sirius Red染色檢測肺組織膠原沉積;免疫組化分析CTSK、SNX9等蛋白在肺組織中的表達與定位;原子力顯微鏡檢測肺組織硬度,評估纖維化程度。
10. 臨床樣本分析:收集PF患者及健康對照的肺組織、支氣管肺泡灌洗液(BALF)和血清樣本,通過ELISA測定血清和BALF中CTSK及谷氨酰胺水平;結合生存分析(Kaplan-Meier)評估CTSK與患者預后的相關性。
11. 轉錄組數據分析:利用公開RNA-seq數據集(GSE124685、GSE70866)分析CTSK家族成員在PF患者中的表達差異及與生存的關聯。
三、主要研究結果
1、CTSK在PF進展過程中的積累
通過蛋白質組學分析PQ誘導ALI小鼠模型的肺組織發現,包括CTSK在內的組織蛋白酶家族成員(如CTSA、CTSB等)在ALI進展過程中(第1、3、7天)逐漸上調,其中CTSK 的積累最為顯著。而在博來霉素(BLM)誘導的PF小鼠肺組織中,CTSK的mRNA水平較對照組顯著升高,且通過免疫組化和Western blot在蛋白水平得到驗證。同時,CTSK的表達在BLM誘導后逐漸增加,于第21天達到峰值,與模型中嚴重PF的發生時間一致。另外,基于BLM誘導PF小鼠的單細胞測序數據(GSE111664),CTSK在巨噬細胞和成纖維細胞中特異性上調,且成纖維細胞數量顯著增加。此外, PF患者的單細胞測序數據(GSE136831)顯示,成纖維細胞中CTSK也呈高表達;在經TGF-β1處理以模擬成纖維細胞-肌成纖維細胞轉化(FMT)的人成纖維細胞系(MRC-5)中,CTSK在mRNA和蛋白水平均顯著上調。
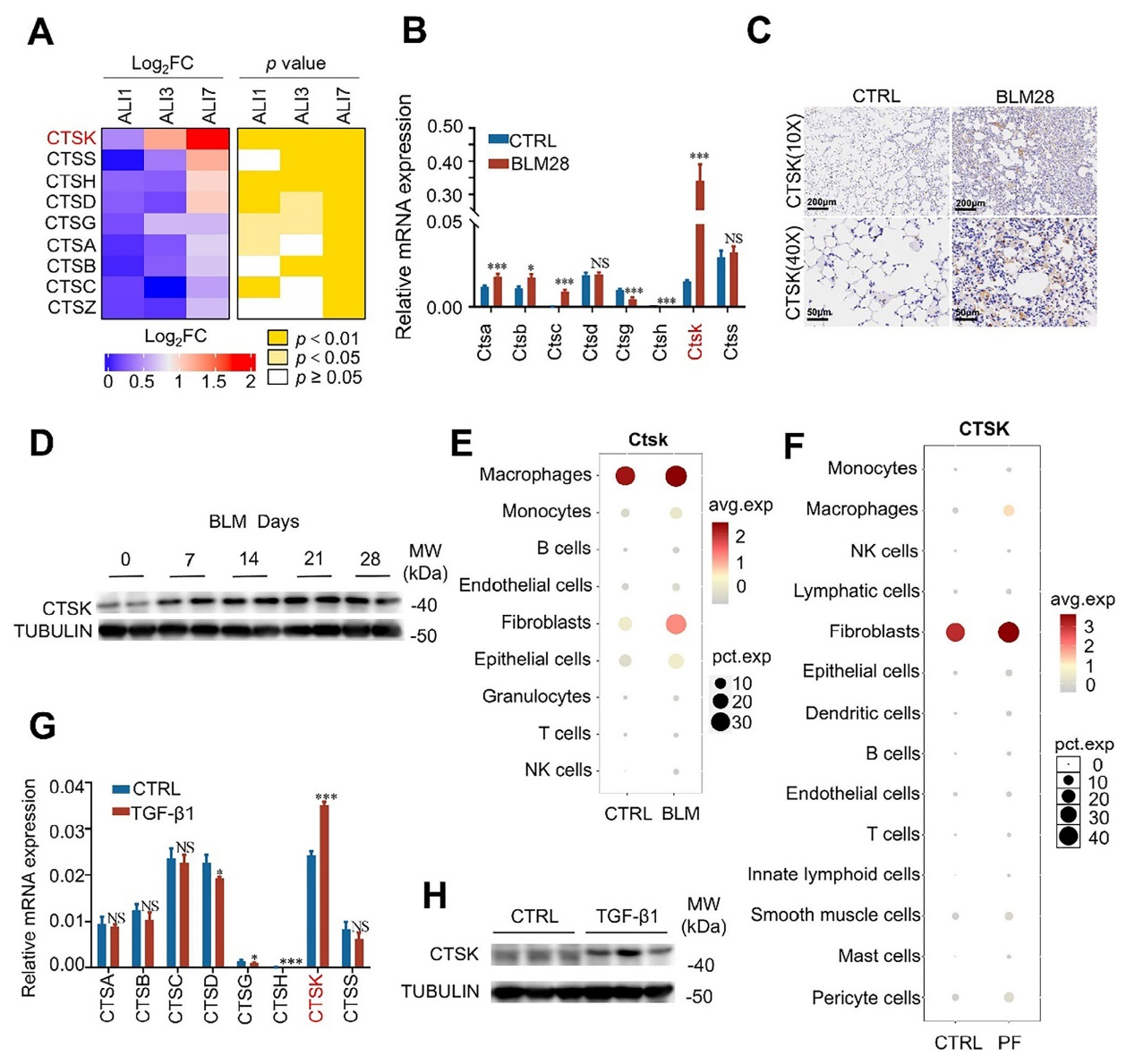
圖1、CTSK在嚴重肺損傷及PF進展過程中在成纖維細胞中富集
2、CTSK對成纖維細胞膠原蛋白合成及肺纖維化PF進程的影響
對PF小鼠的單細胞測序數據(GSE132771)分析顯示,CTSK陽性成纖維細胞中膠原蛋白相關基因(如 Col1a1、Col1a2、Col3a1)的表達顯著高于CTSK陰性成纖維細胞,提示CTSK可能促進膠原蛋白合成。同時,在TGF-β1誘導的人成纖維細胞系(MRC-5)中,高濃度(0.1 μg/mL)的重組CTSK(rCTSK)經24小時處理后,可特異性上調COL1A1的表達,而對 COL1A2、COL3A1及α-SMA的表達無顯著影響,且該結果在原代小鼠肺成纖維細胞中得到驗證。另外,通過蛋白酶體抑制劑(MG132)和蛋白質合成抑制劑(環己酰亞胺)實驗發現,rCTSK通過促進COL1A1的合成(而非抑制其降解)來增加其蛋白水平,且對α-SMA的表達無影響。在BLM誘導的PF小鼠模型中,施用rCTSK后,肺組織中COL1A1的表達和羥脯氨酸含量顯著增加,Masson三色染色和Sirius Red染色顯示膠原沉積和膠原纖維增多,原子力顯微鏡(AFM)證實肺組織硬度增加;同時,纖維化標志物mRNA水平也顯著升高。rCTSK對纖維化進程的時序影響研究結果顯示,BLM誘導后,在第7天施用rCTSK可顯著上調第14天和第21天肺組織中CTSK和Col1a1的蛋白表達,且這些時間點的血清 CTSK水平也明顯升高,表明過量CTSK在肺損傷后會加劇PF進展。綜上,圖2的結果證實CTSK可通過促進成纖維細胞中膠原蛋白(尤其是COL1A1)的合成,加劇肺纖維化的嚴重程度。
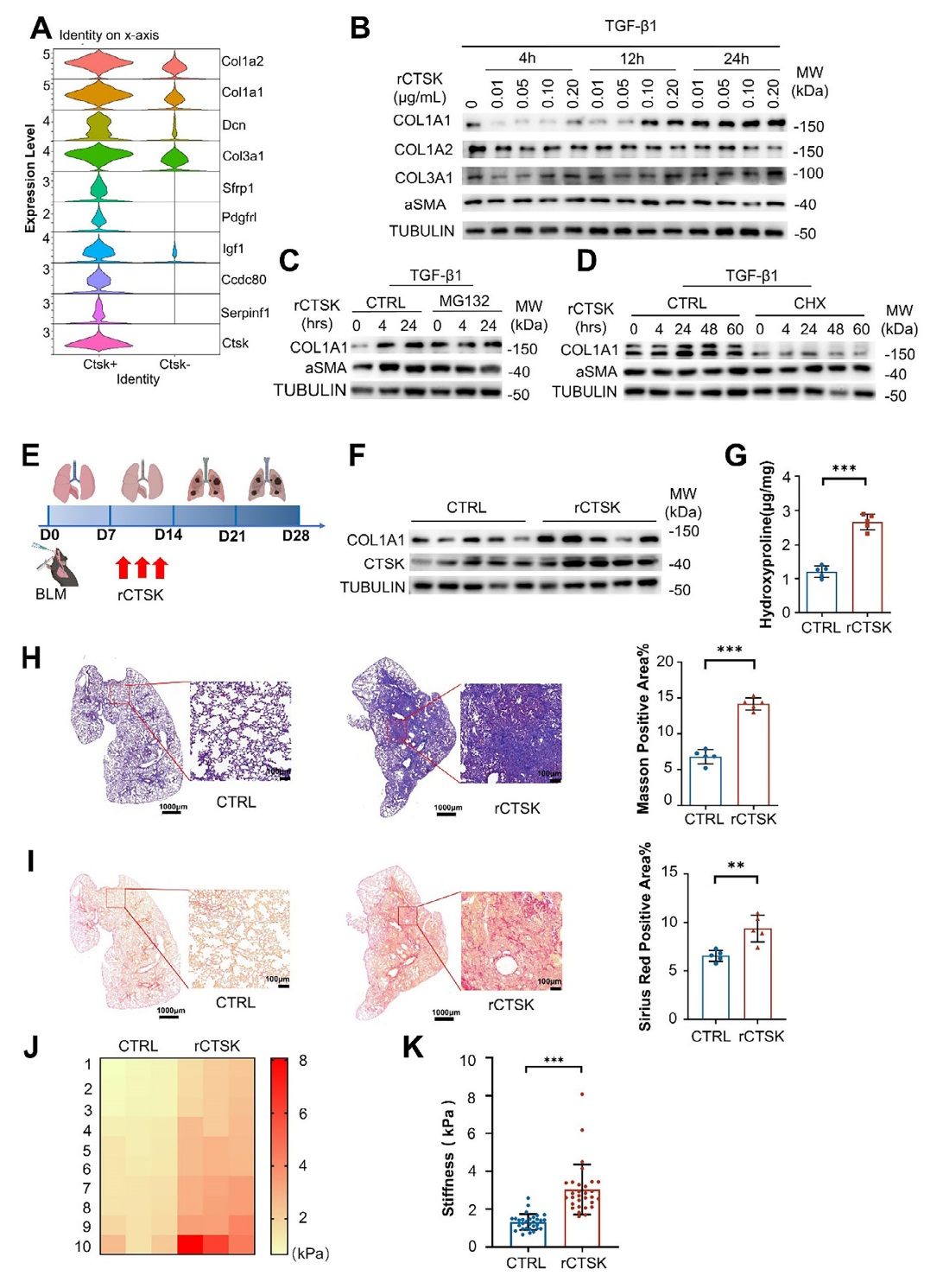
圖2、CTSK可通過促進成纖維細胞中膠原蛋白的合成加劇肺纖維化的嚴重程度
3、CTSK與SNX9的相互作用及其對成纖維細胞膠原蛋白合成的調控機制

圖3、CTSK與SNX9結合促進成纖維細胞中膠原蛋白合成進而加劇肺纖維化進程
通過免疫共沉淀(Co-IP)實驗驗證,在經TGF-β1和重組CTSK(rCTSK)處理的人成纖維細胞系(MRC-5)中,CTSK與SNX9存在直接相互作用,且隨著處理時間延長,COL1A1 和SNX9的積累增加。AlphaFold3預測結合界面顯示,CTSK的LYS220/CYS221殘基與 SNX9的TRP39殘基是相互作用的關鍵位點,丙氨酸突變實驗進一步證實了這些位點的重要性;免疫熒光顯示CTSK與SNX9主要在細胞質和核膜共定位,提示CTSK可能通過與 SNX9結合被內吞。在MRC-5細胞中敲低SNX9后,TGF-β1誘導的COL1A1表達降低,且 rCTSK無法再上調COL1A1的產生,表明SNX9是CTSK促進膠原蛋白合成的關鍵介質。另外,使用網格蛋白介導的內吞作用(CME)抑制劑Pitstop2處理MRC-5細胞后,CTSK的細胞內積累減少,同時COL1A1的表達也顯著降低,證實內吞過程在CTSK調控膠原蛋白合成中不可少的。此外,體內驗證實驗表面,在BLM誘導的肺纖維化小鼠模型中,聯合施用rCTSK和Pitstop2后,與單獨施用rCTSK的小鼠相比,肺組織中羥脯氨酸含量、COL1A1 和CTSK蛋白水平顯著降低,Masson和Sirius Red染色顯示膠原沉積減少,纖維化標志物的mRNA水平也下降,表明阻斷SNX9介導的內吞可緩解CTSK加劇的肺纖維化。該部分結果揭示CTSK通過與SNX9結合并經內吞作用,促進成纖維細胞中膠原蛋白合成,進而加劇肺纖維化進程。
4、CTSK通過TGF-β1-SMAD3信號通路促進膠原蛋白合成的機制研究
在經TGF-β1和重組CTSK(rCTSK)處理的人成纖維細胞系(MRC-5)中,SMAD3的磷酸化水平隨處理時間延長顯著升高,且與COL1A1的表達增加同步;在BLM誘導的肺纖維化小鼠模型中,施用rCTSK后,肺組織中磷酸化SMAD3(p-SMAD3)、COL1A1、SNX9和 CTSK的蛋白水平均上調。而在MRC-5細胞中敲低SNX9后,rCTSK增強的胞質p-SMAD3水平顯著降低,且核內p-SMAD3的積累也大幅減少;免疫熒光實驗進一步證實,SNX9敲低后,p-SMAD3與SNX9的共定位減少,表明SNX9是CTSK激活SMAD3的關鍵介質。免疫共沉淀(Co-IP)實驗顯示,CTSK、SNX9與p-SMAD3之間存在關聯,提示三者可能形成復合物共同參與信號傳遞。此外,使用SMAD3抑制劑SIS3處理MRC-5細胞后,無論是基礎狀態還是rCTSK誘導的COL1A1表達均被顯著抑制,同時細胞內CTSK水平也降低,證實SMAD3激活是CTSK促進膠原蛋白合成的必要環節。圖4的結果揭示CTSK通過 SNX9介導的內吞作用,激活TGF-β1誘導的SMAD3磷酸化,進而促進成纖維細胞中膠原蛋白的合成。
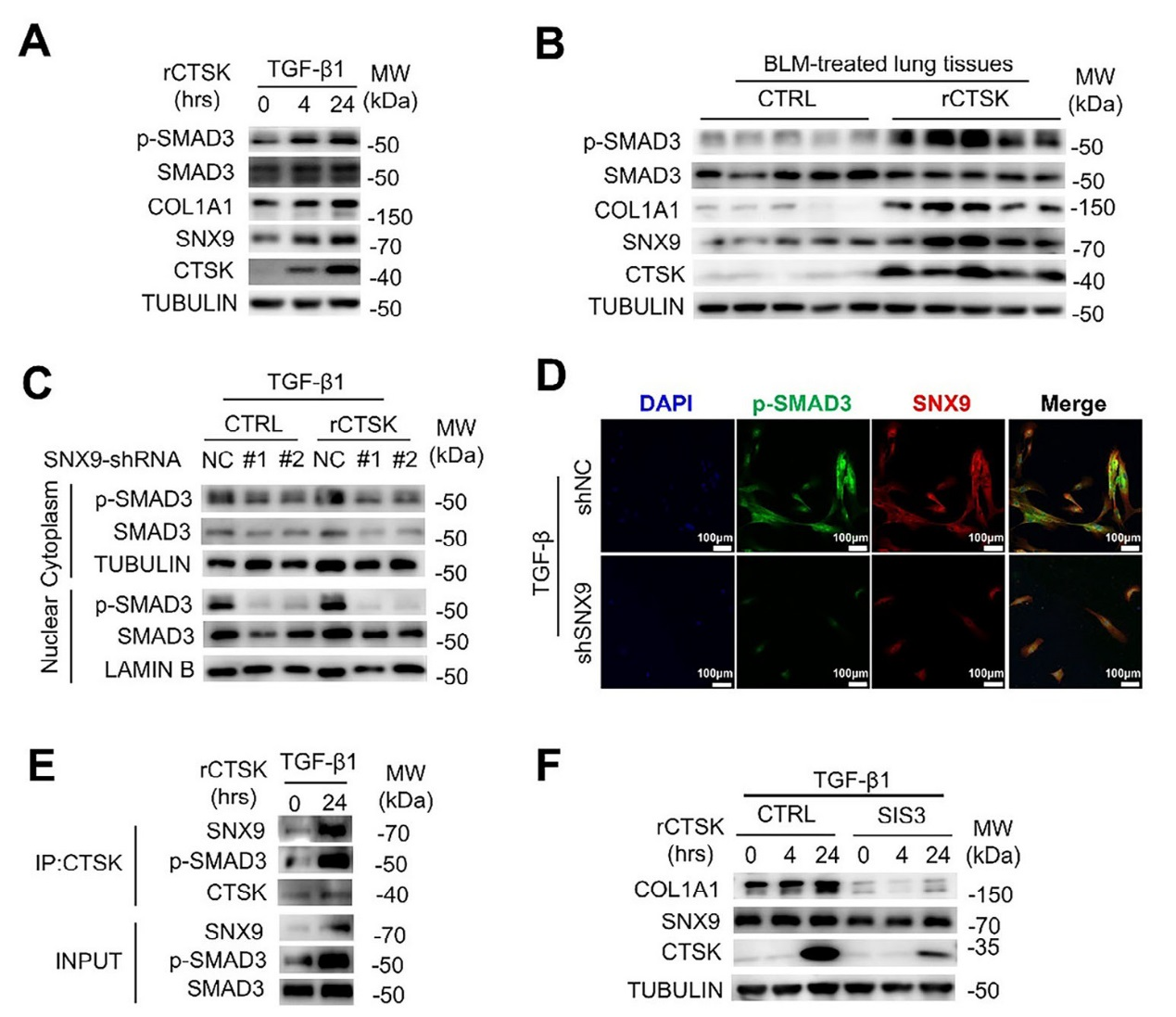
圖4、CTSK通過SNX9介導的內吞作用,激活TGF-β1誘導的SMAD3磷酸化,進而促進成纖維細胞中膠原蛋白的合成
5、CTSK對谷氨酰胺代謝的調控及其在膠原蛋白合成中的作用
對PF患者的單細胞測序數據(GSE136831)分析顯示,CTSK陽性成纖維細胞中谷氨酸顯著富集,提示CTSK可能與谷氨酰胺代謝相關。同時,對經TGF-β1和重組CTSK(rCTSK)處理的MRC-5細胞進行非靶向代謝組學分析,KEGG和MSEA富集分析表明,rCTSK顯著激活包括谷氨酰胺代謝在內的氨基酸相關通路,其中谷氨酰胺代謝通路因參與膠原蛋白合成所需的脯氨酸和甘氨酸生成而成為關鍵通路。靶向代謝組學檢測顯示,rCTSK處理后,TGF-β1誘導的MRC-5細胞中谷氨酰胺、谷氨酸和脯氨酸水平顯著升高,其中谷氨酰胺和脯氨酸的增加更為明顯。谷氨酰胺酶1(GLS1)是谷氨酰胺代謝的關鍵酶,rCTSK可上調TGF-β1誘導的MRC-5細胞中GLS1的mRNA和蛋白表達及酶活性;而敲低SNX9后,rCTSK對GLS1的上調作用被顯著抑制,谷氨酰胺水平也隨之降低,表明CTSK通過SNX9促進GLS1介導的谷氨酰胺代謝。綜上,圖5的結果揭示CTSK通過SNX9增強GLS1的表達和活性,激活谷氨酰胺代謝通路,為膠原蛋白合成提供原料,進而促進肺纖維化進程。
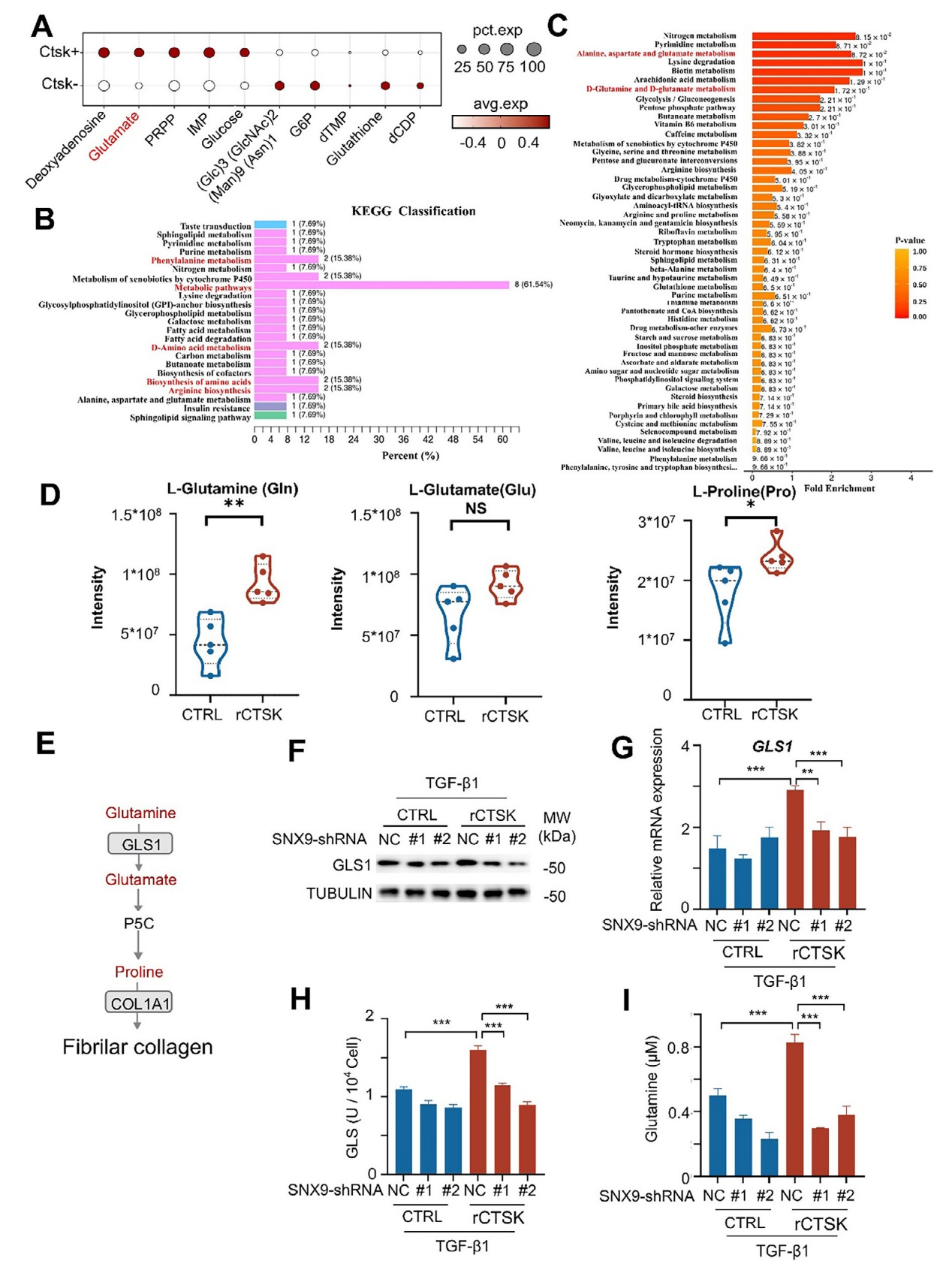
圖5、CTSK高表達促進谷氨酰胺代謝
6、CTSK通過谷氨酰胺代謝促進成纖維細胞膠原蛋白合成的機制研究
在不含谷氨酰胺的培養基中,經TGF-β1和重組CTSK(rCTSK)處理的人成纖維細胞系(MRC-5)中,COL1A1的表達顯著降低,而α-SMA水平不受影響;反之,在培養基中額外添加谷氨酰胺可顯著增加COL1A1的產生,表明谷氨酰胺是CTSK促進膠原蛋白合成的關鍵原料。
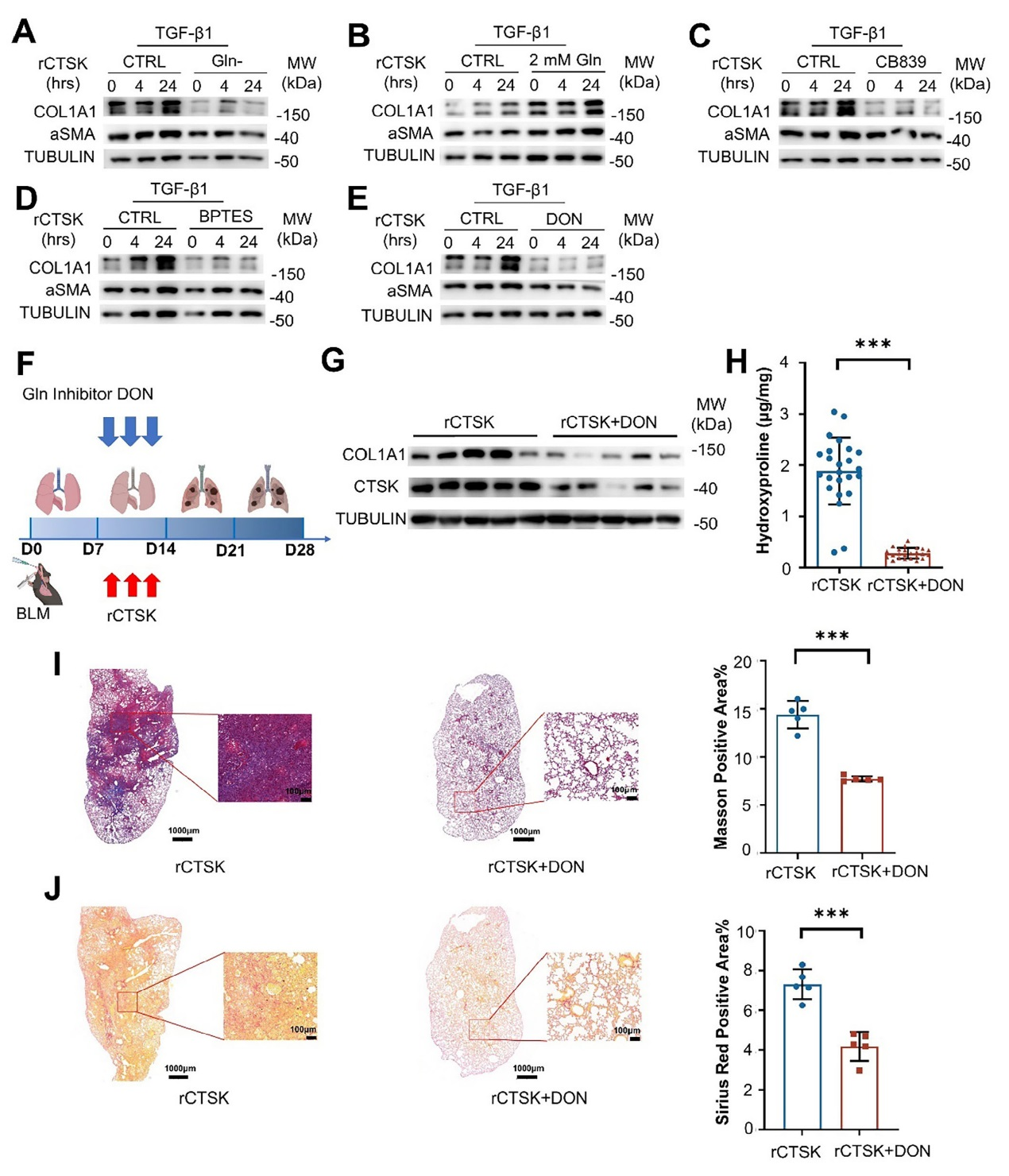
圖6、CTSK觸發的谷氨酰胺代謝促進成纖維細胞膠原合成
另外,使用谷氨酰胺酶(GLS1)抑制劑(CB839、BPTES、DON)處理MRC-5細胞后,rCTSK 誘導的COL1A1表達被顯著抑制,但α-SMA水平未受影響,證實GLS1介導的谷氨酰胺代謝在CTSK促進膠原蛋白合成中起核心作用。而在BLM誘導的肺纖維化小鼠模型中,聯合施用rCTSK和DON(GLS1 抑制劑)后,與單獨施用rCTSK的小鼠相比,肺組織中COL1A1的表達、羥脯氨酸含量顯著降低,Masson三色染色和Sirius Red染色顯示膠原沉積減少,纖維化標志物的mRNA水平也下降,表明抑制谷氨酰胺代謝可緩解CTSK加劇的肺纖維化。該部分的結果證實CTSK通過激活谷氨酰胺代謝通路促進成纖維細胞膠原蛋白合成,而抑制該代謝通路可有效減輕肺纖維化程度。
7、CTSK表達與谷氨酰胺水平及PF患者的預后相關性分析
對PF患者的RNA-seq數據(GSE124685)分析顯示,包括CTSK在內的組織蛋白酶家族成員在肺組織中顯著上調,其中CTSK的表達水平最高。免疫組化和Western blot驗證顯示,PF患者肺組織中CTSK、SNX9、GLS1及磷酸化SMAD3(p-SMAD3)的表達均顯著高于健康對照;多重免疫熒光進一步證實這些分子在纖維化肺組織中存在共定位,提示它們在PF 發病中可能協同作用。此外,基于PF患者的RNA-seq數據(GSE70866)的生存分析顯示,CTSK高表達患者的生存率顯著低于低表達患者,表明CTSK可作為PF預后的潛在標志物。另外,PF患者的支氣管肺泡灌洗液(BALF)和血清中CTSK水平顯著高于健康對照及非 PF的急性呼吸窘迫綜合征(ARDS)患者;且BALF和血清中CTSK水平高的患者死亡率顯著升高。同時,PF患者血清中谷氨酰胺水平顯著升高,且與CTSK水平呈正相關,進一步支持CTSK通過調控谷氨酰胺代謝影響PF進展的機制在臨床中的相關性。結果表明,CTSK及其相關分子在PF患者中異常上調,血清CTSK水平可作為 PF 預后評估的潛在標志物,且其與谷氨酰胺水平的相關性為臨床干預提供了方向。

圖7、患有肺纖維化(PF)的急性呼吸窘迫綜合征(ARDS)患者中,外周組織蛋白酶 K(CTSK)含量與谷氨酰胺水平相關,且提示預后不良
四、全文結論
研究表明,組織蛋白酶 K(CTSK)在肺纖維化(PF)中通過雙重機制發揮作用:早期降解細胞外基質,后期過量積累則與分選連接蛋白 9(SNX9)結合被內吞,激活TGF-β1-SMAD3 通路,上調谷氨酰胺酶 1(GLS1),促進谷氨酰胺代謝和膠原蛋白合成,加劇PF。臨床數據顯示,PF患者血清CTSK水平與谷氨酰胺水平正相關,且高CTSK預示不良預后。綜上,CTSK和谷氨酰胺可作為PF預后標志物及治療靶點。
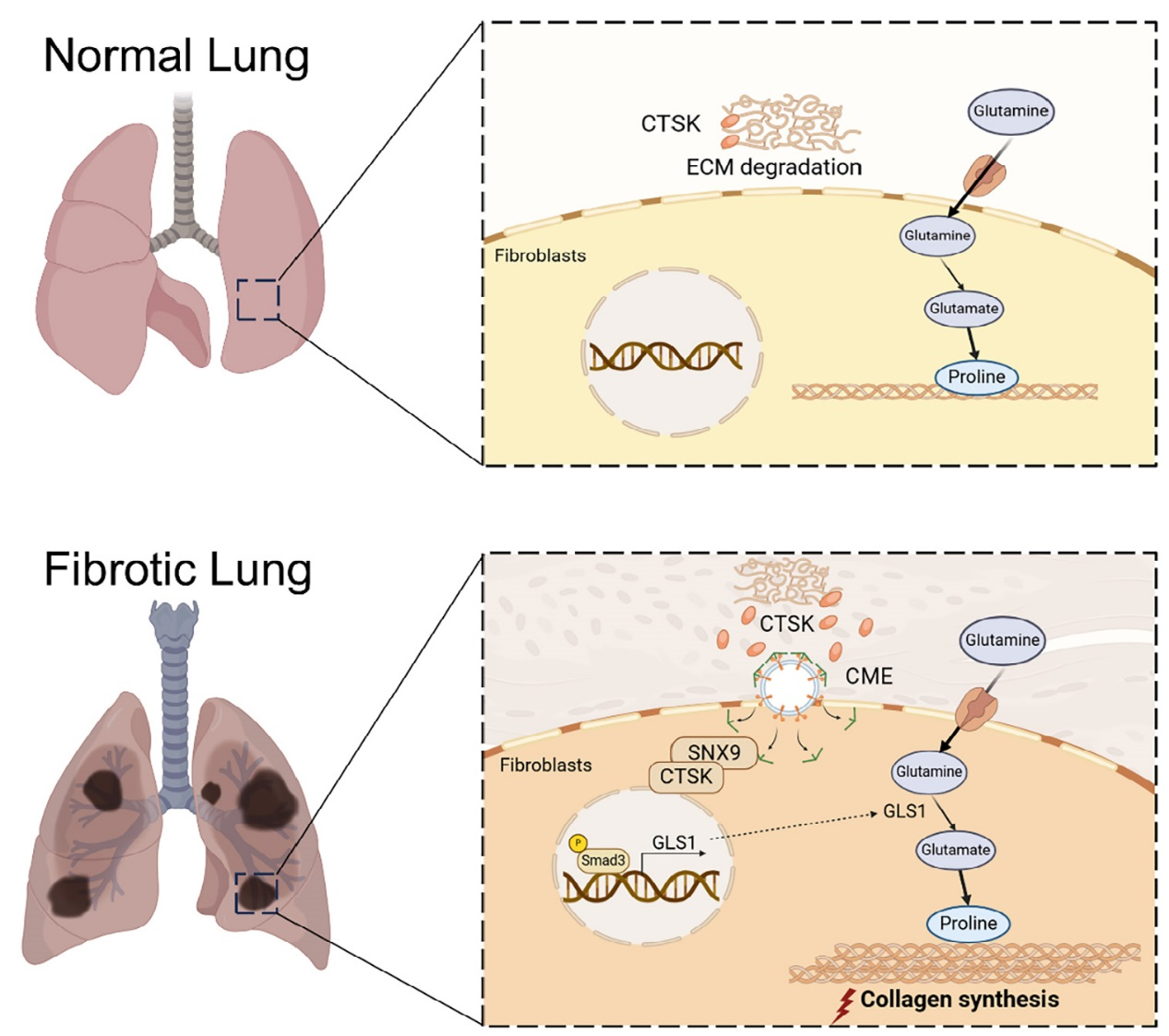
圖8、流程圖
參考文獻:
Chen M, Meng X, Zhu Y, Wang D, Wang M, Wang Z, Tian X, Zhang J, Yue Z, Yang Z, Wang R. Cathepsin K Aggravates Pulmonary Fibrosis Through Promoting Fibroblast Glutamine Metabolism and Collagen Synthesis. Adv Sci (Weinh). 2025 Jul 3:e13017. doi: 10.1002/advs.202413017. Epub ahead of print. PMID: 40605618.

 當前位置:
當前位置:
